CHARMM: MD Simulation of RNA and DNA
CHARMM: MD Simulation of RNA and DNA
Objective and Overview
This tutorial provides an overview of the CHARMM commands required to build and simulate either a DNA or RNA duplex
in explicit solvent.
In addition, an input file provides the appropriate CHARMM commands for converting RNA, which is the default nucleic acid in CHARMM, to DNA.
Note, all remaining steps, including solvation, minimization, and simulation, are the same for both DNA and RNA.
An image of a duplex is shown on the right
|
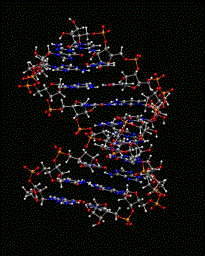
|
Generation of RNA and DNA Duplexes
Initial Input Coordinates from a PDB File Downloaded from the Protein Data Bank
A nucleic acid duplex (PDB Identifer: 1RRR)
is initially downloaded from the Protein Data Bank.
Then, the PDB coordinates are converted to CRD format acceptable to CHARMM.
Topology and parameter files used for the calculations are the additive all-atom
CHARMM Force Field Files which are located in the tutorial directory.
Building RNA and DNA
RNA
Once the initial CRD file is created for the RNA structure, it is then used in the CHARMM script file, helix_rna_gen.inp,
from which a PSF file and new CRD file are obtained.
DNA
For the CHARMM force field, the RNA form of nucleic acids is the default.
Thus, for the building a DNA duplex structure, the CHARMM commands are almost the same as for the RNA duplex, but there is an additional step
of removing the 2'OH moieties, as shown in the CHARMM script, helix_dna_gen.inp.
Using the PDB File Directly in CHARMM
Small changes to the CHARMM scripts allow the use of a PDB file as input for the coordinates:
|
CHARMM CRD Input Coordinates
open unit 20 read form name rr_1_noh.pdb.crd
read coor card unit 20
|
PDB Input Coordinates
rename segid a select segid rna1 end
rename segid b select segid rna2 end
open unit 20 read form name rr_1.pdb
read coor pdb unit 20 resid
rename segid rna1 a select segid a end
rename segid rna2 b select segid b end
|
|
For PDB file input, the segment names are renamed for each chain to match those in the rr_1.pdb file, and then
changes them back so the segments have the same names in the later CHARMM scripts.
|
MD Simulation
Solvation and Neutralization
Once the RNA (or DNA) duplex is constructed, it is then solvated by overlaying an equilibrated water box, followed by deletion of
all water molecules that are in too close-contact with RNA.
The system is then checked to insure that it is electronically neutral. If the system is not neutral, Na+
ions are added at random positions by first removing an H2O molecule, subsequently replacing H2O with an Na+ ion, and then ensuring that
it is not in too-close contact with either the RNA or other solvent molecules at that position.
In general, ions can be added or deleted to achieve a neutral system.
Note that the present approach yields a system with only minimal salt.
If a higher salt concentration is required it is suggested that, more Na+ and Cl- ions be added
to obtain the desired ionic strength. An image of the solvated and neutralized system is shown to the right.
charmm < helix_dna_solvate.inp > helix_dna_solvate.log
charmm < helix_dna_neutralize.inp > helix_dna_neutralize.log
|
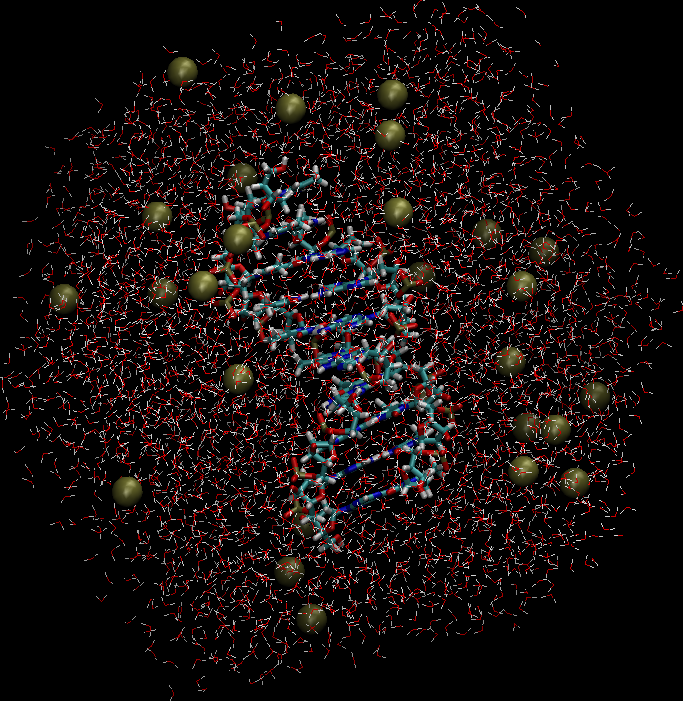
|
Heating, Equilibration, and Production
The remainder of the CHARMM scripts involve equilibration of the system around the RNA followed by the production MD simulation.
During equilbration, the coordinates of the DNA/RNA are harmonically constrained and a
minimization is performed. This is followed by a NVT simulation to allow the solvent and ions to equilibrate around the
nucleic acid. The final coordinates from the equilibration are used for the production simulation,
The production simulation involves an NPT simulation with long-range electrostatics treated via the Particle-Mesh Ewald method.
Note, that the trajectory is run, such that a series of trajectory files, along with the associated restart are created.
This allows the simulation to be readily restarted and facilitates handling of the files during analysis.
Analysis
Two simple analysis CHARMM scripts to determine the RMS Differences as a function of time (rms.inp) and the base pairing energy
as a function of time (bp_interactions.inp) are included in the analysis subdirectory.
Then, run the CHARMM script files in the analysis directory:
cd analysis
charmm < rms.inp > rms.log
charmm < bp_interactions.inp > bp_interactions.log