This tutorial provides an introduction to NAMD, and some of its basic capabilities, including the steps of a molecular dynamics simulation, including preparation, minimization, equilibration, production, and finally, analysis of a system. Also, various options and output of the NAMD simulation program are described. In addition, typical simulation techniques are introduced, and the analysis of properties for molecular systems is also illustrated.
A typical Molecular Dynamics simulation using NAMD includes the following stages:
To build the initial structure, one needs the following:
In this exercise, the coordinates for deca-alanine are given by the file alanine_original.pdb in PDB format.
The file deca-alanine.pgn constructs an initial deca-alanine structure and contains the following statements:
package require psfgen
resetpsf
topology toppar_c36_feb16/top_all36_prot.rtf
pdbalias residue HOH TIP3
pdbalias residue HIS HSE
pdbalias atom ILE CD1 CD
segment seg0 {
first ACE
last CT3
pdb alanine.pdb
}
coordpdb alanine.pdb seg0
guesscoord
writepsf output/deca-alanine.psf
writepdb output/deca-alanine.pdb
exit
The above file can be executed with the either of the following commands:
psfgen alanine.pgn > alanine.log
or
vmd -dispdev text -e alanine.pgn > alanine.log
where the psfgen program is part of both the NAMD and vmd distributions which can be invoked through the module command.
The psf and pdb files generated by the psfgen step can then be combined into a NAMD configuration file ( alanine_min.conf) which minimizes the isolated protein whose contents are shown below:
# namd configuration file to minimize deca-alanine protein chain # molecular system structure output/deca-alanine.psf coordinates output/deca-alanine.pdb temperature 300 # Output Control set outputname output/deca-alanine-minimize outputName $outputname ########################### ## SIMULATION PARAMETERS ## ########################### # CHARMM Force Field Parameters paraTypeCharmm on parameters toppar_c36_feb16/par_all36_prot.prm parameters toppar_c36_feb16/par_all36_lipid.prm parameters toppar_c36_feb16/par_all36_na.prm parameters toppar_c36_feb16/par_all36_carb.prm parameters toppar_c36_feb16/par_all36_cgenff.prm parameters toppar_c36_feb16/toppar_water_ions.str # Simulation space partitioning exclude scaled1-4 1-4scaling 1.0 cutoff 12. switching on switchdist 10. pairlistdist 13.5 #Minimize for a number of steps minimize 100 namd2 deca-alanine-minimize.namd > deca-alanine-minimize.log
vmd -dispdev text -e solvate_add_ions.tclaccomplishes these steps. The contents of the solvate_add_ions.tcl script file are given below:
# Load PSF and binary coordinate file
mol new output/deca-alanine.psf type psf
mol addfile output/deca-alanine-minimize.coor type namdbin
#measure the minimum and maximum values of coordinates
set coords [atomselect top all]
measure minmax $coords
#center the alanine helix
set a [measure center $coords]
set aminus [vecscale -1 $a]
$coords moveby $aminus
#see if it worked
measure center $coords
# Write out the centered alanine helix
$coords writepdb output/deca-alanine-centered.pdb
#solvate the recentered alanine helix with a water box
#new psf and pdb files are written
package require solvate
solvate output/deca-alanine.psf output/deca-alanine-centered.pdb -t 10 -o output/deca-alanine-centered-solvated
#check coordinates to see if the solvated alanine helix box is centered
set coords [atomselect top all]
measure center $coords
#Now add ions.
#new psf and pdb files are written
package require autoionize
autoionize -psf output/deca-alanine-centered-solvated.psf -pdb output/deca-alanine-centered-solvated.pdb \
-sc 0.15 -cation POT -anion CLA -o output/deca-alanine-solvated-ions
#recenter the alanine helix after solvation and adding ions
set coords [atomselect top "not protein"]
measure minmax $coords
set a [measure center $coords]
set aminus [vecscale -1 $a]
$coords moveby $aminus
set coords [atomselect top protein]
measure minmax $coords
set a [measure center $coords]
set aminus [vecscale -1 $a]
$coords moveby $aminus
#analyze the geometric components of the solvated and ionized system
set coords [atomselect top all]
measure center $coords
#measure the minimum and maximum values of coordinates
#to get an idea of the cell parameters
measure minmax $coords
quit
NAMD configuration scripts for heating + equilibration and production are similar, but there are several differences as well. Below is shown only the NAMD production script, deca-alanine-solvated-ions-production.namd:
######################################################################
# Production Dynamics of alanine in a Water Box #
######################################################################
# Molecular system
structure output/deca-alanine-solvated-ions.psf
# Output Control
set outputname output/deca-alanine-solvated-ions-production5
outputName $outputname
binaryoutput no
restartfreq 500 ;# 500steps = every 1ps
dcdfreq 500
velDCDfile $outputname.vel.dcd
velDCDfreq 500
xstFreq 500
dcdunitcell yes
outputEnergies 500
outputPressure 500
# Input Control
set inputname output/deca-alanine-solvated-ions-equilibrate
binaryrestart yes
bincoordinates $inputname.restart.coor
binvelocities $inputname.restart.vel
extendedSystem $inputname.restart.xsc
coordinates output/deca-alanine-solvated-ions-equilibrate.coor
firsttimestep 0
###########################
## SIMULATION PARAMETERS ##
###########################
# CHARMM Force Field Parameters
paraTypeCharmm on
parameters toppar_c36_feb16/par_all36_prot.prm
parameters toppar_c36_feb16/par_all36_lipid.prm
parameters toppar_c36_feb16/par_all36_na.prm
parameters toppar_c36_feb16/par_all36_carb.prm
parameters toppar_c36_feb16/par_all36_cgenff.prm
parameters toppar_c36_feb16/toppar_water_ions.str
# Simulation space partitioning
exclude scaled1-4
1-4scaling 1.0
cutoff 12.
switching on
switchdist 10.
pairlistdist 13.5
# Integrator Parameters
timestep 2.0 ;# 2fs/step
rigidBonds all ;# needed for 2fs steps
nonbondedFreq 1
fullElectFrequency 2
stepspercycle 10
# Constant Temperature and Pressure Control - NPT Dynamics
# langevin piston is required, which provides the pressure control
langevinPiston on
langevinPistonTarget 1.01325
langevinPistonPeriod 200
langevinPistonDecay 50
langevinPistonTemp 300
useGroupPressure yes ;# needed for rigidBonds
useFlexibleCell yes
useConstantArea yes
# Periodic Boundary Conditions
cellBasisVector1 32.0 0.00 0.00
cellBasisVector2 0.00 32.0 0.00
cellBasisVector3 0.00 0.00 32.0
cellOrigin 0.00 0.00 0.00
wrapAll on
# PME for full-system periodic electrostatics
PME yes
PMEGridSizeX 32
PMEGridSizeY 32
PMEGridSizeZ 32
run 10000; #run sets the number of timesteps over which
#to run MD equilibration or production
To run this script, the following command is used:
namd2 +p24 deca-alanine-solvated-ions-production.namd > deca-alanine-solvated-ions-production.logNotice, that the "+p24" option is added to the namd2 command for the production run, allowing execution on one node with 24 SMP processors (cores). Other numbers of cores can be used depending on the Linux node. Increasing the number of cores executing in parallel is very desirable for long executing production simulations. The result will be an increase in turn around time and a concomitant decrease in elapsed execution time.
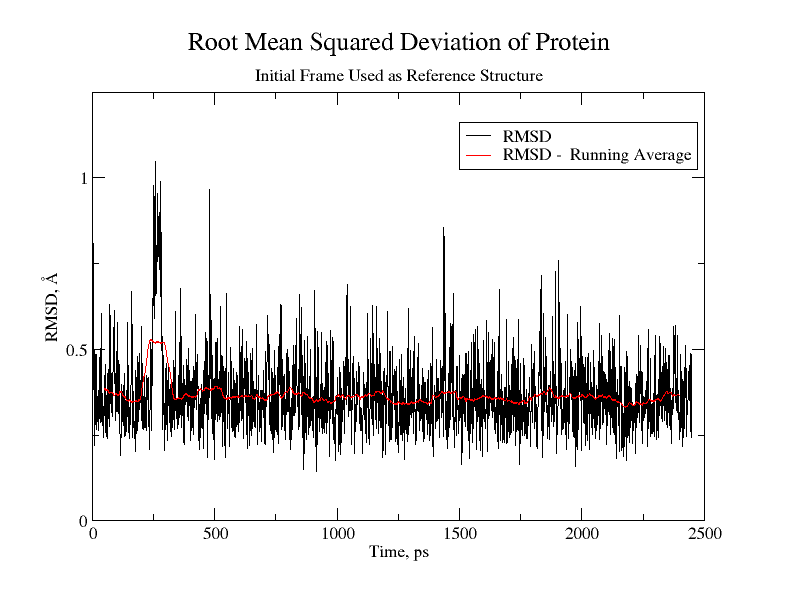
When one or more trajectory files are obtained from production runs, then it possible to perform trajectory analysis of the solvated protein system. One important property for analysis involves RMSD, which is the root mean square deviation between the coordinates from a Molecular Dynamics simulation and the coordinates from an experimental protein structure.
The Root Mean Squared Difference (RMSD) is the difference between individual trajectory frames of coordinates with respect to the coordinates of a reference crystal structure of a protein or the first trajectory frame. The RMSD tells us something about the dissimilarity between the structures - 1 Å RMSD is barely visible to the eye, while an RMSD of 10 Å is leading to a significantly different structure.
Thus, the behavior of the protein from an initial starting structure or crystal reference structure, can be calculated with the following tcl script (rmsd.tcl) :
source bigdcd.tcl
# This script computes the RMS distance between each frame in a sequence of
# .xyz files and a reference pdb file. The script is set up for
# batch processing, i.e. it waits for the analysis to complete
# and then exits.
#
set outfile [open "rmsd.dat" w]
proc myrmsd { frame } {
global outfile ref sel all
$all move [measure fit $sel $ref]
puts $outfile "$frame [measure rmsd $sel $ref]"
}
set mol [mol new test_ions.psf type psf waitfor all]
mol addfile test_ions.pdb type pdb waitfor all
set all [atomselect $mol all]
set ref [atomselect $mol "name CA" frame 0]
set sel [atomselect $mol "name CA"]
bigdcd myrmsd dcd test_ions_production.dcd
bigdcd_wait
quit
The command:
vmd -dispdev text -e rmsd.tclcreates a file containing two columns of data, including the time versus RMSD value. This can be used as one measure of whether or not the system has equilibrated, and a graph of the RMSD of the protein versus time is shown below:
Many other properties of the solvated protein system can be obtained by using the tcl scripts located in the exercise directory.